Research Interests
Our research interest focuses on characterizing the relation between structure, interactions, and dynamics of molecular systems in liquid phase at the atomistic/microscopic scale. In particular, the systems that we study are polymers, designer solvents, and electrolytes. The goal of our research is to help to elucidate the relation between structure and macroscopic properties by gaining detailed information regarding dynamic and interactions at the molecular level. This information is vital for the development and tailoring of these materials in existent or new applications.
Metal ion batteries are the energy storage technology of choice in most of the portable electronics. However, the link between the macroscopic and microscopic structure and dynamics of these metal ion electrolytes is still missing.This project is focused on understanding the solvation of metal cations in organic solvents from the perspective of both the solvent and cation and anion, and aiming at finding how its solvation is affected by the interrelations between the solvent molecular structure and the chemical identity of the lithium salt. The solvation of metal cations is hard to investigate because of its wide range of possible coordination numbers, its competitive and/or cooperative solvation in solvent mixtures, the overlap between the different solvation sheaths, the rapid solvent exchange, and the formation of ion-pairs. Thus, we use ultrafast IR spectroscopy to disentangle the different species present in solution.
Selected references:
Fulfer KD, Kuroda DG, "A comparison of the solvation structure and dynamics of the lithium ion in linear organic carbonates with different alkyl chain lengths", Physical Chemistry Chemical Physics, 2017, 19, 25140-25150.
Fulfer KD, Kuroda DG, "Solvation Structure and Dynamics of the Lithium Ion in Organic Carbonate-Based Electrolytes: A Time-Dependent Infrared Spectroscopy Study",Journal of Physical Chemistry C, 2016, 120, 24011-24022.
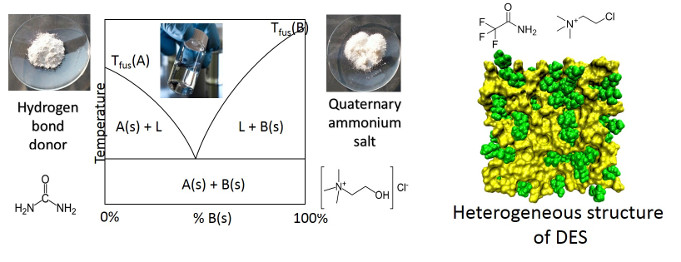
Deep eutectic solvents (DES) are a new type of designer solvent formed binary mixture of an ionic compound with a hydrogen-bond donor. Due to their simple preparation and tunable properties, DES are an inexpensive alternative to conventional solvents. However, the molecular structure and interactions between the DES ingredients giving rise to their properties are lacking. This project aims at characterizing the DES molecular structure and interactions among components to rationalize the physico-chemical properties of these materials. However, the structure and dynamics of solvents and their solutions are difficult to characterize because of the low symmetry and fast exchange of the solvent molecular arrangements. Thus, we explore the structure using IR spectroscopies in combination with theoretical methods.
 Selected references:
Selected references:
Yaowen Cui and Daniel G Kuroda, "Evidence of Molecular Heterogeneities in Amide Based Deep Eutectic Solvents", J. Phys. Chem. A, 2018, 122, 1185-1193.
Cui Y, Fulfer KD, Ma J, Weldeghiorghis TK, Kuroda DG, "Solvation Dynamics of an Ionic Probed in Choline Chloride-based Deep Eutectic Solvents", Physical Chemistry Chemical Physics, 2016, 18, 31471-31479.
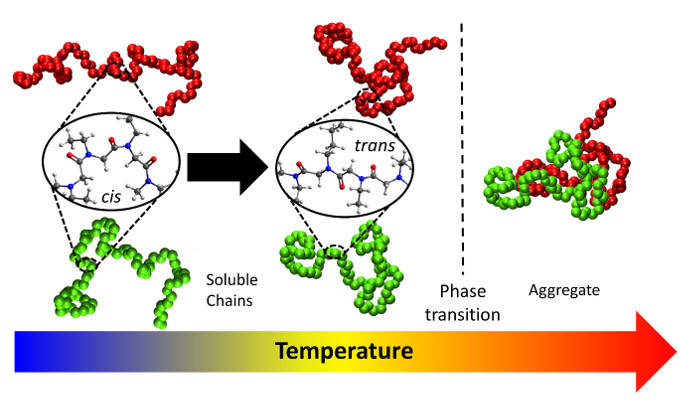
Thermoresponsive polymers are polymers that exhibit severe changes in solubility with temperature. This research project concentrates on studying the molecular rearrangements of the polymer and its solvation at different temperatures under equilibrium and non-equilibrium conditions for different thermoresponsive polymers. In particular, this project aims at characterizing the change in molecular structure and dynamics of both the solvent and the polymer below, at, and above transition temperature using an array of different experimental techniques, such 2DIR, NMR, etc.
 Selected references:
Selected references:
Ma J, Xuan S, Guerin AA, Yu T, Zhang D, Kuroda DG, "Unusual molecular mechanism behind the thermal response of polypeptoids in aqueous solutions", Physical Chemistry Chemical Physics, 2017, 19, 10878-10888.
Methodologies
To investigate these projects, we are using a comprehensive approach that combines well-established experimental methodologies, such as Two-dimensional infrared (2DIR) spectroscopy, and computer modeling, including molecular dynamics simulations and quantum chemical calculations.
Vibrational spectroscopy measures the molecular absorption due to the promotion to an excited vibrational state. The frequencies of the absorption bands have associated vibrational modes which can be well localized modes, which allow one to probe fairly confined regions of the molecule.Moreover, it has been shown that in liquids the molecular vibrational modes are very susceptible to the composition and structure of the solvation shell. Thus, vibrational spectroscopy is used to characterize the chemical state of the molecular components in solutions. While it is fairly straightforward to assign vibrational transitions to vibrational modes, the presence of combination modes makes their assignment challenging. Therefore, other vibrational techniques are be used together to disentangle the IR spectrum. Further, vibrational spectroscopy can be used to quantify chemical species in the solution at different concentrations and temperatures to determine the equilibrium constant and energetics of process, like hydrogen bond making and breaking.Steady state IR spectroscopies allows us to measure the speciation of a molecule in solution, but it does not provide us with the dynamics of the process affecting the probed molecules. In contrast, ultrafast vibrational spectroscopies can achieve femtosecond (10-15s) time resolution which permits us to probe structure and dynamics of chemical and physical processes on the same time scale as atomic motions. Thus, one can use high-order nonlinear IR spectroscopies, 2DIR or pump-probe, to monitor specific vibrational modes of a molecule as a function of time. Monitoring the time evolution of vibrational modes allows us to gain molecular-level insights into the system and its dynamics in condensed phase.

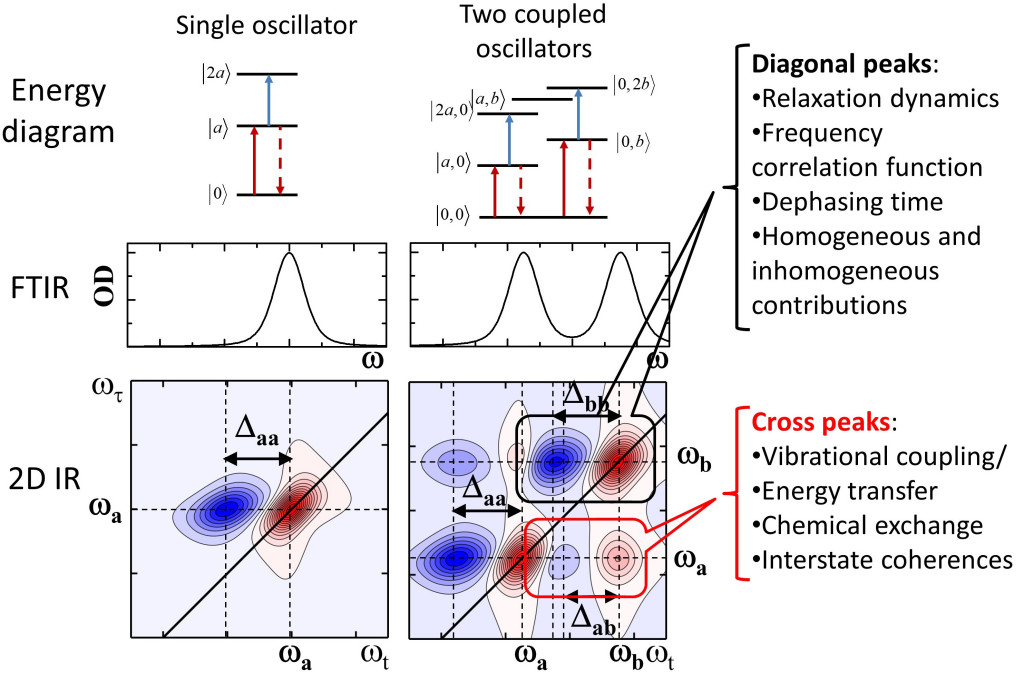
Two dimensional IR (2DIR) spectroscopy consists of measuring the response of a molecular system after it interacts with three ultrafast IR laser pulses. While the first two pulses tag the vibrational mode to be followed, the third pulse reads its temporal evolution by generating a non-linear signal.
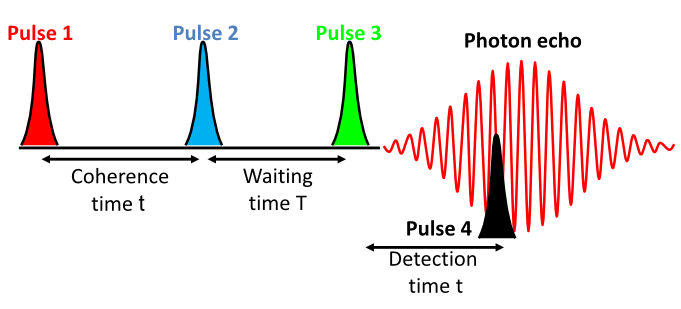 The signal measured as a function of the three time intervals is usually represented
as the 2DIR spectrum. The detailed analysis and modeling of the 2DIR spectra, which
includes line shapes, cross-peaks, and other time dependent features, is used to directly
monitor molecular events (such as making/breaking hydrogen bonds) as they happen.
The signal measured as a function of the three time intervals is usually represented
as the 2DIR spectrum. The detailed analysis and modeling of the 2DIR spectra, which
includes line shapes, cross-peaks, and other time dependent features, is used to directly
monitor molecular events (such as making/breaking hydrogen bonds) as they happen.
 A state of the art IR optical setup is used for these 2DIR experiments.
A state of the art IR optical setup is used for these 2DIR experiments.

Pump-probe spectroscopy is another non-linear spectroscopy used to measure ultrafast processes. This spectroscopy is based on "probing" the response of the sample generated by the pump pulse. In particular, by monitoring the probe signal as a function of the time delay between the pump and probe, one can obtain information about molecular processes generated by the vibraitonal excitation of the sample, such as vibrational population lifetimes.
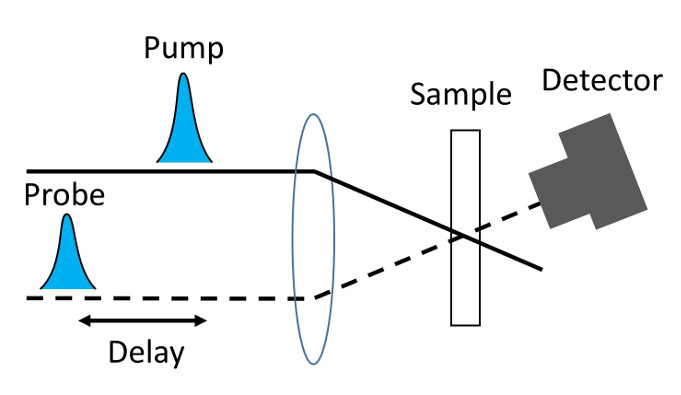 In addition, one can measure the polarization dependence of the probe signal as function
of the time delay to obtain the time dependence anisotropy of the sample, which contains
the dynamics of molecular processes, like molecular reorientation.
In addition, one can measure the polarization dependence of the probe signal as function
of the time delay to obtain the time dependence anisotropy of the sample, which contains
the dynamics of molecular processes, like molecular reorientation.
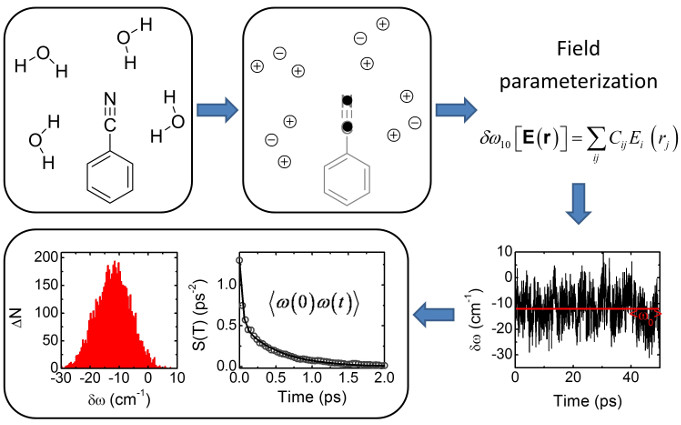
In addition, we model our experimental findings using both first-principle and classical molecular dynamics simulations to understand the complex 2DIR spectrum and to gain an atomistic detail of the processes giving rise to the 2DIR spectral features and their time evolution. An example of these events is the hydration shell reorganization in aqueous solutions which usually occurs on a sub-picosecond (>10-12 seconds) time scale.
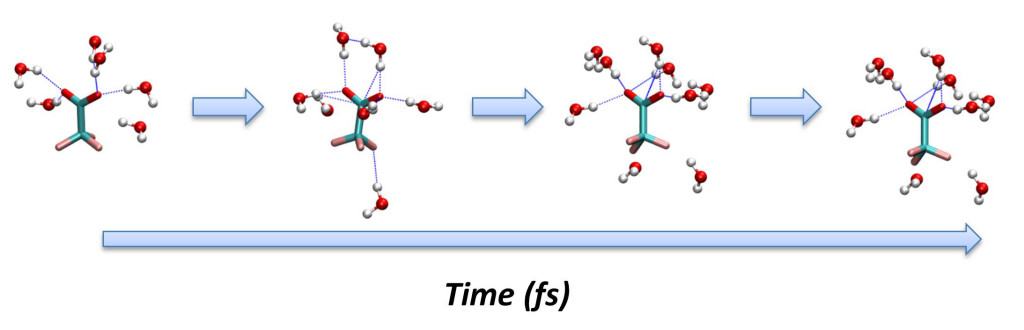 Thus, we use can use the dynamics from molecular dynamics simulations to obtain the
solvation shell reorganization motions and translate them into frequency fluctuations
via a DFT vibrational map. Once, the instantaneous frequencies are computed the experimental
observables, such as the FFCF, can be calculated.
Thus, we use can use the dynamics from molecular dynamics simulations to obtain the
solvation shell reorganization motions and translate them into frequency fluctuations
via a DFT vibrational map. Once, the instantaneous frequencies are computed the experimental
observables, such as the FFCF, can be calculated.