Cardiac fibroblasts and cardiac fibrosis
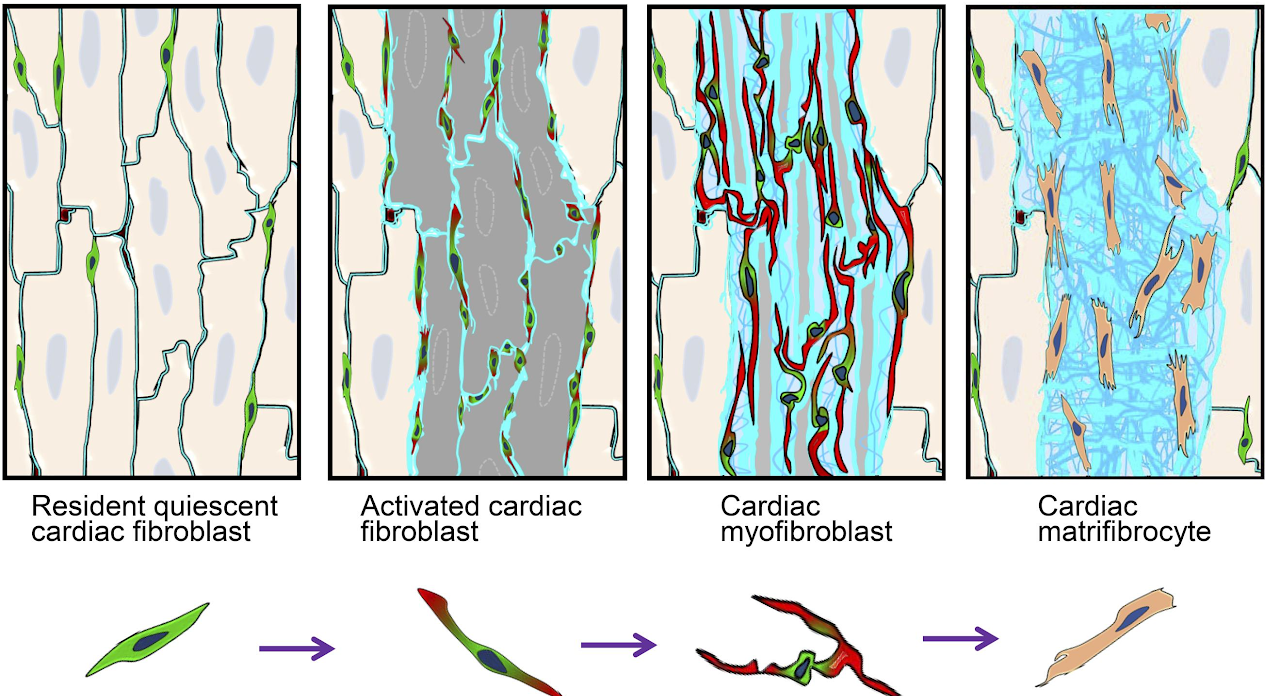
Cardiac fibroblasts are a group of tissue resident mesenchymal cells. These cells stay largely quiescent in the interstitial area between cardiomyocytes under normal condition. However, when heart is injured such as after myocardial infarction (MI), these cells quickly start to proliferate and then differentiate to myofibroblasts expressing high levels of extracellular matrix (ECM) proteins and highly organized α smooth muscle actin (αSMA). Previous research mainly focused on myofibroblasts due to their obvious importance in most-injury tissue repair. And it is believed that myofibroblasts either dedifferentiate back to fibroblast or undergo apoptosis when the tissue repair is finished. However, we recently found that cardiac fibroblasts persist in the infarct scar long term after MI likely due to the nonregenerative nature of heart. These persistent cardiac fibroblasts do not express αSMA, the hallmark of myofibroblasts, but express moderate levels of some typical heart ECM proteins and some unique proteins that are usually expressed in bone and cartilage. These cells also lack the ability to proliferate in response to additional stimuli. We, therefore, named these cells matrifibrocytes. The unique properties of these cells suggest an important role of them in maintaining the post-MI infarct stability. Using a multiomic approach, combining RNAseq and ATACseq, we characterized the gene expression and chromatin remodeling during the post-MI activation and differentation process of cardiac fibroblasts.
Articles
- Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart (JCI.org)
- The landscape of accessible chromatin in quiescent cardiac fibroblasts and cardiac fibroblasts activated after myocardial infarction (tandfonline.com)
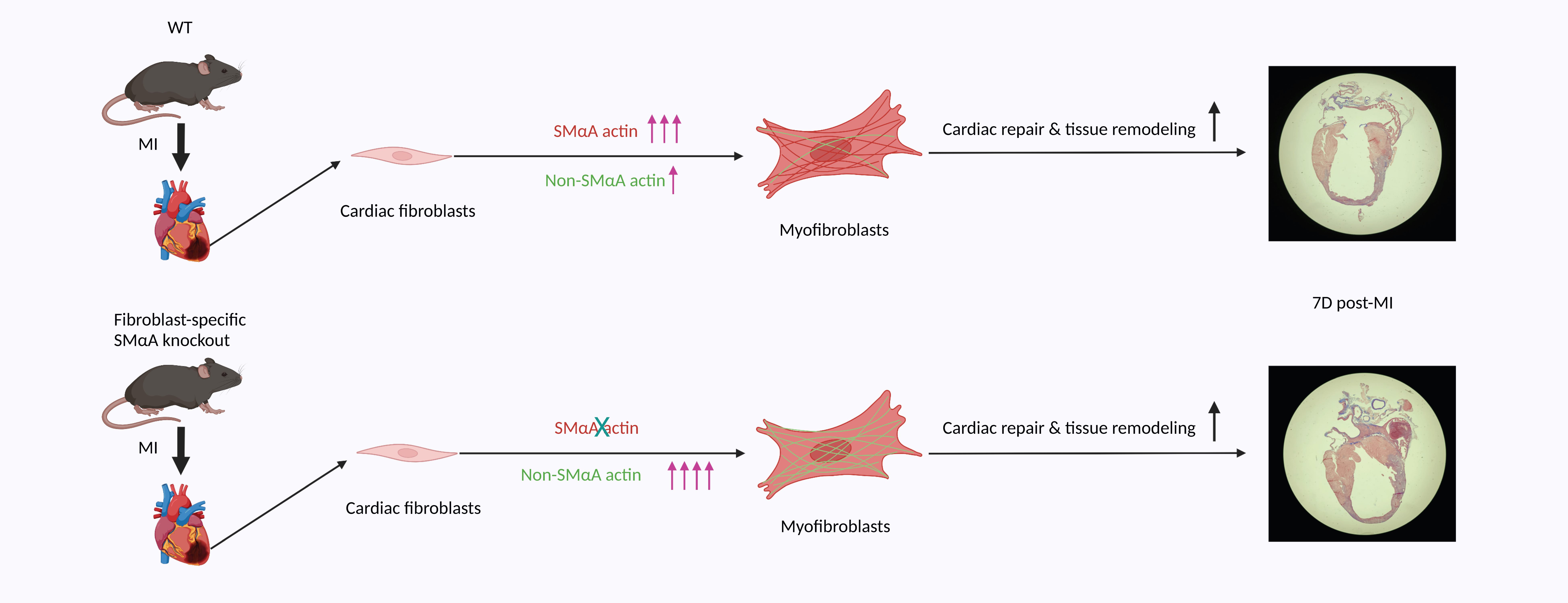
In response to myocardial infarction (MI), quiescent cardiac fibroblasts differentiate into myofibroblasts mediating tissue repair. One of the most widely accepted markers of myofibroblast differentiation is the expression of Acta2 which encodes smooth muscle alpha-actin (SMαA) that is assembled into stress fibers. However, the requirement of Acta2/SMαA in the myofibroblast differentiation of cardiac fibroblasts and its role in post-MI cardiac repair remained unknown. To answer these questions, we generated a tamoxifen-inducible cardiac fibroblast-specific Acta2 knockout mouse line. Surprisingly, mice that lacked Acta2 in cardiac fibroblasts had a normal post-MI survival rate. Moreover, Acta2 deletion did not affect the function or histology of infarcted hearts. No difference was detected in the proliferation, migration, or contractility between WT and Acta2-null cardiac myofibroblasts. Acta2-null cardiac myofibroblasts had a normal total filamentous actin level and total actin level. Acta2 deletion caused a significant compensatory increase in the transcription level of non-Acta2 actin isoforms, especially Actg2 and Acta1. Moreover, in myofibroblasts, the transcription levels of cytoplasmic actin isoforms were significantly higher than those of muscle actin isoforms. In addition, we found that myocardin-related transcription factor-A is critical for myofibroblast differentiation but is not required for the compensatory effects of non-Acta2 isoforms. In conclusion, the Acta2 deletion does not prevent the myofibroblast differentiation of cardiac fibroblasts or affect the post-MI cardiac repair, and the increased expression and stress fiber formation of non-SMαA actin isoforms and the functional redundancy between actin isoforms are able to compensate for the loss of Acta2 in cardiac myofibroblasts.
Article
- Loss of Acta2 in cardiac fibroblasts does not prevent the myofibroblast differentiation or affect the cardiac repair after myocardial infarction (ScienceDirect.com)