Lopata Research Group
Louisiana State University
Department of Chemistry
We use computer simulations to predict how electrons move in molecules at the attosecond time scale.
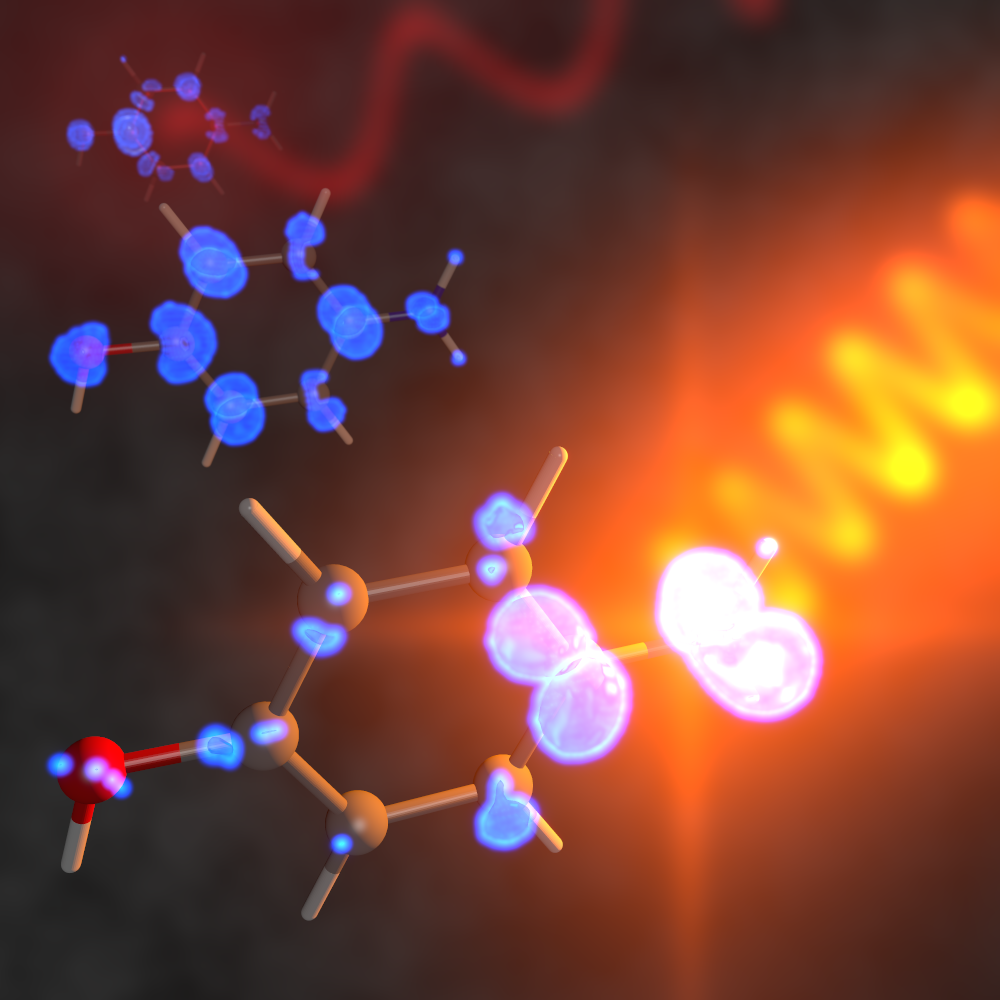
Simulated attosecond electron density motion in an aminophenol molecule following excitation with a ultraviolet laser pulse, and an artist's representation of an X-ray pulse probing the density around the nitrogen atom.J. Chem. Theory Comput. 16, 4470 (2020)
When forced out of equilibrium, electrons in molecules respond at an extraordinarily fast time scale, typically a few hundred attoseconds. One attosecond corresponds to a billionth of a billionth (10-18) of a second, making these dynamics faster than even the time it takes for atoms to move.
The movement of electrons is integral to processes such as light harvesting, photochemistry, and radiation damage. Experiments at the frontier of ultrafast science are increasingly capable of measuring these processes, but much is currently unknown about initial electron behavior, especially those resulting from interaction with laser pulses.
Attosecond electron dynamics, which are governed by the laws of quantum mechanics, are best thought of as a flow of probability density. To understand how these density flows occur, we use quantum chemistry computer simulations to determine which molecules support dynamics, how it occurs, and to assist in interpret corresponding experiments.
These virtual experiments allow us to unravel the "attochemistry" mechanisms of electron dynamics.
Our current research interests include:
- Charge migration
- Interactions of molecules and solids with intense laser fields
- X-ray absorption and scattering for probing attosecond processes
We use real-time electronic structure approaches, primarily real-time time-dependent density functional theory (RT-TDDFT) with localized basis sets and hybrid/range-separated hybrids. This includes non-Hermitian versions for strong-field ionization, and variations modified for core-level spectroscopy.